Michael Rosen, an admired and respected investor for decades, penned this excellent blog that summarizes the state of protein structure prediction and how it has evolved since 1994 to today. So well written, below we offer his article in its entirety, along with our heartfelt thanks.

There are twenty amino acids in the human body. Amino acids are the chemical links that make up proteins. Proteins perform all sorts of essential tasks. Hemoglobin, for example, is the protein molecule in red blood cells that carries oxygen from the lungs to the body’s tissues and returns carbon dioxide from the tissues back to the lungs. Keratin is the type of protein that makes up your hair, skin, and nails. The spike (S) protein plays a key role in the receptor recognition and cell membrane fusion process in SARS-CoV-2.
There are approximately 30 trillion cells in the human body. Each cell contains between one billion and three billion proteins. How can a mere 20 amino acids make billions of proteins?
The answer is how each protein folds on itself to form its final shape. We can see this through X-ray crystallography, which sends electromagnetic radiation to interact with molecular crystals that reveal each atom of a molecule. This is great, but X-ray crystallography is time-consuming and very expensive.
The great molecular biologist, Cyrus Levinthal, estimated that there are 10300 possible configurations of a typical protein. Brute calculation of each variation is impossible: it would take longer than the age of the universe (almost 14 billion years) to identify each combination. Another approach is required if we hope to know the structure of proteins.
Every two years since 1994, scientists have gathered for a competition to see who could create an algorithm that could accurately predict the shape of proteins using only a list of its amino acids. The competition is called the Critical Assessment of Protein Structure Prediction, and the answer is no one. Participants are given 43 proteins to model, and the best programs were able to get two or three right. Until 2018, when DeepMind’s AlphaFold program successfully predicted 25 of the 43 proteins (the second-place program got three right). The competition was held again last month, and DeepMind’s newer version, AlphaFold2, achieved an astonishing accuracy of 92.4%.
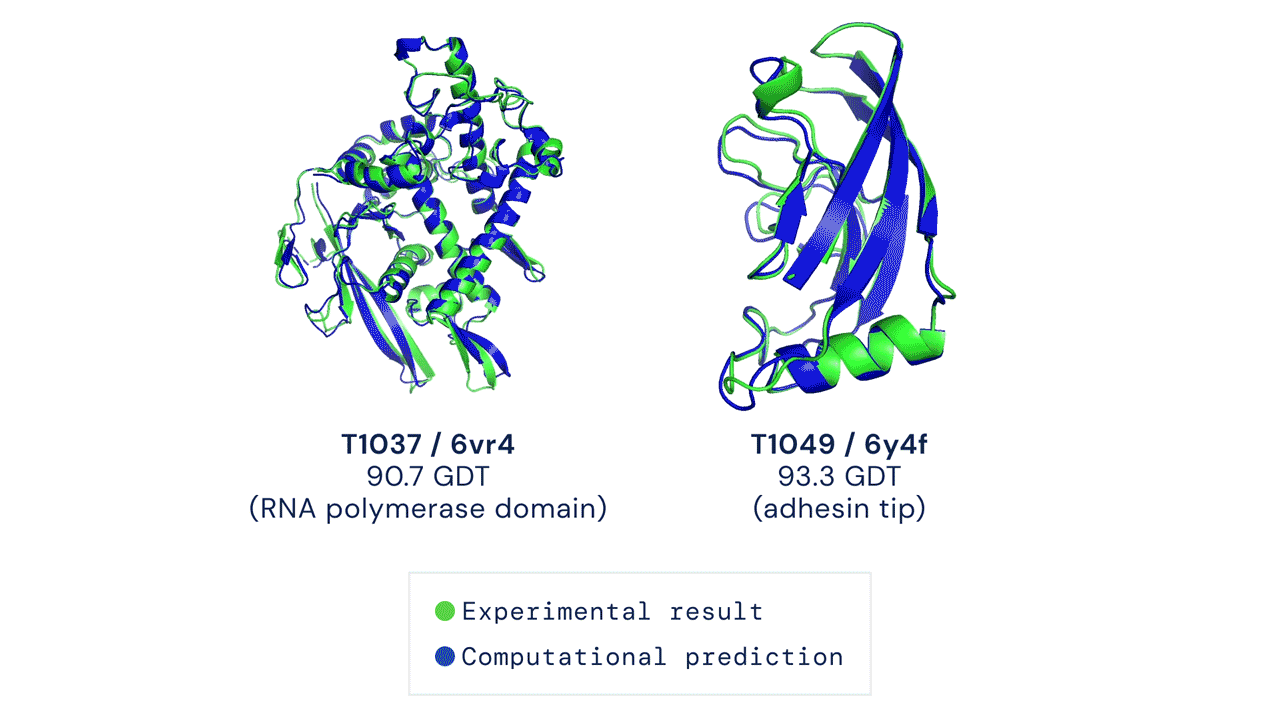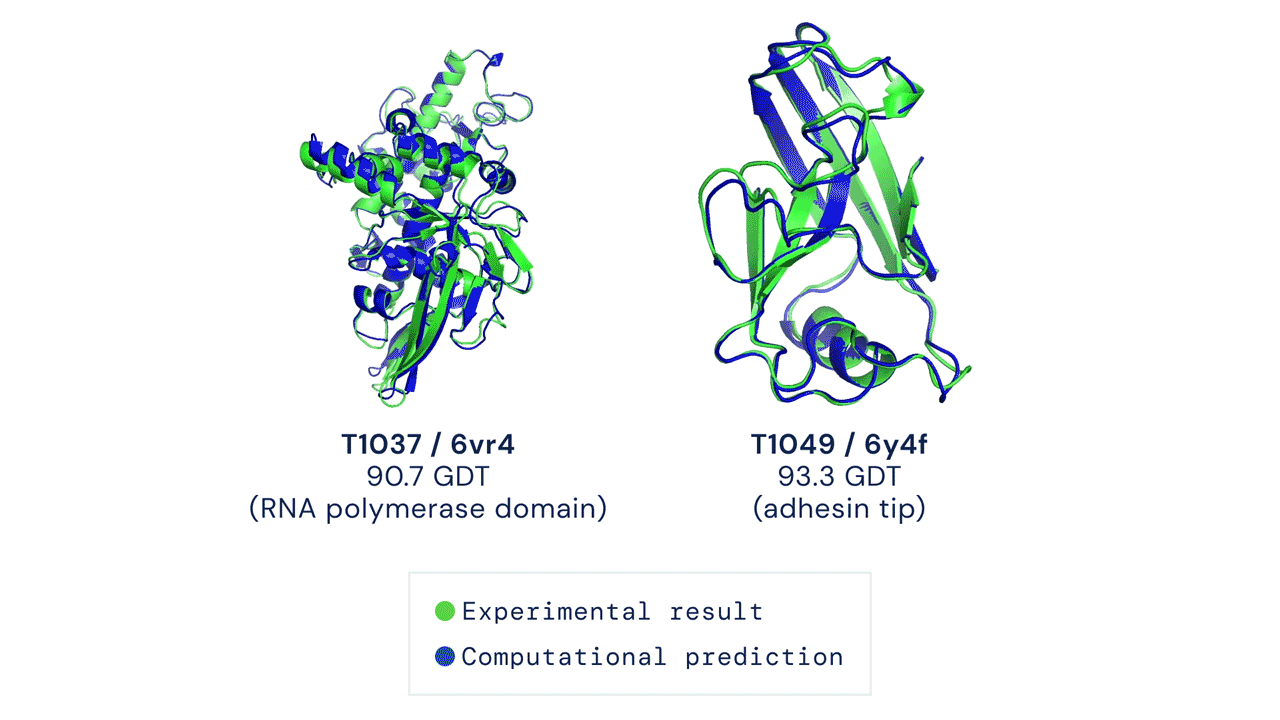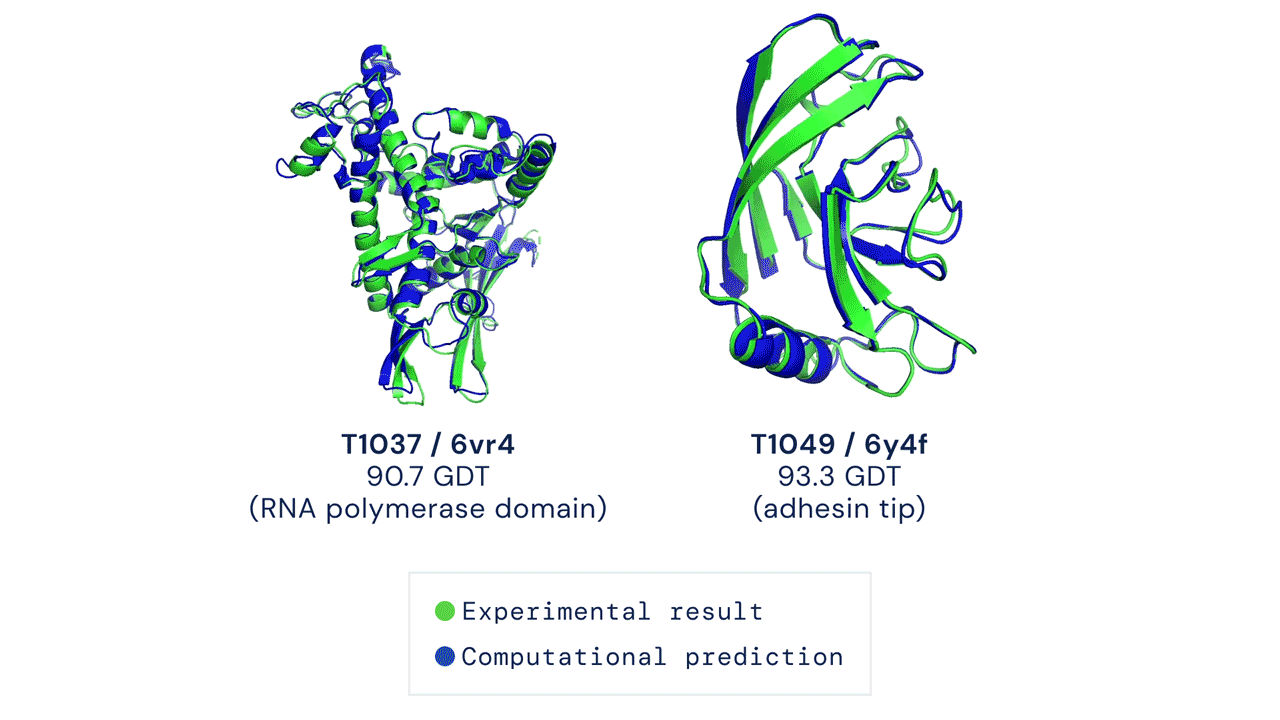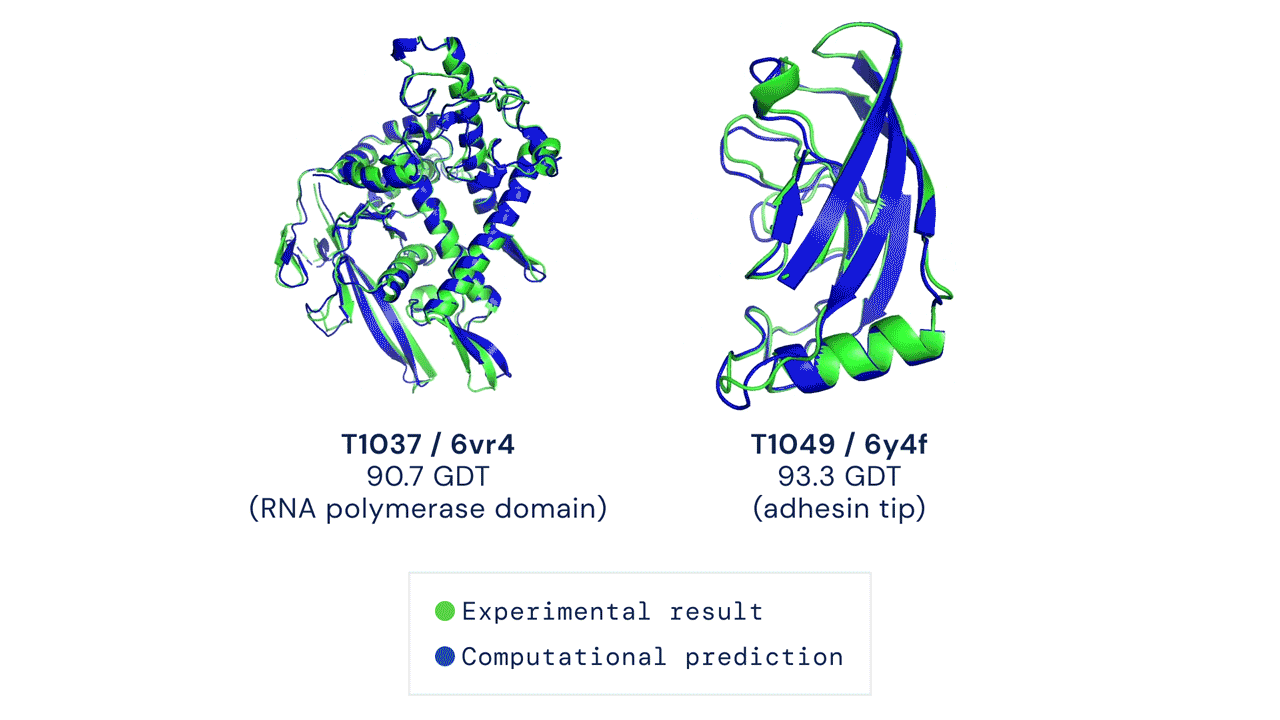
A folded protein can be thought of as a “spatial graph”, and DeepMind (based in the UK and owned by Alphabet) built a neural network that uses evolutionarily related sequences, multiple sequence alignment (MSA), and a representation of amino acid residue pairs to refine and interpret this graph while reasoning over the implicit graph that it’s building. By iterating this process over a few days, AlphaFold developed strong predictions of the underlying physical structure of the protein and was able to calculate which parts of each predicted protein structure are reliable using an internal confidence measure.
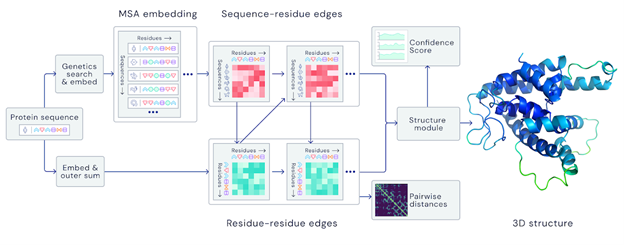
AN OVERVIEW OF THE MAIN NEURAL NETWORK MODEL ARCHITECTURE. THE MODEL OPERATES OVER EVOLUTIONARILY RELATED PROTEIN SEQUENCES AS WELL AS AMINO ACID RESIDUE PAIRS, ITERATIVELY PASSING INFORMATION BETWEEN BOTH REPRESENTATIONS TO GENERATE A STRUCTURE.
This is an extraordinary accomplishment: from a list of the 20 amino acids that comprise a protein, AlphaFold was able to predict the shape of that protein, out of 10300 possibilities, to an accuracy of 92.4%. Earlier this year, AlphaFold predicted several protein structures of the SARS-CoV-2 virus, including ORF3a and another coronavirus protein, ORF8, whose structures were previously unknown. Experimentalists have confirmed the existence of both these structures.
The implications of this achievement cannot be overstated. A misshapen protein is thought to be the cause of Alzheimer’s and many other diseases. If we could only identify the shape of that protein, we could have a chance to correct it. For the first time, with AlphaFold, we now have that chance. We will be better able to determine which drugs are likely to bind to a particular protein and to effectively design proteins to catalyze chemical reactions.
In 2003, the Human Genome Project (and Celera Genomics) successfully mapped the entire human genome. Since then, the Universal Protein database has collected 180 million protein sequences, but only 170,000 have had their structures determined because of the time and expense required to do so. AlphaFold represents an exponential leap forward in being able to determine protein structures, and thus is a huge step toward the effective treatment of diseases.
As investors, we obsess over the vicissitudes of our political discourse (such as it is) as if it were a sporting contest. We scrutinize each economic release and infer the hidden meanings of a central banker’s pronouncements. Most of what fills our working days is noise, and we are easily distracted from the achievements that will profoundly determine our future. AlphaFold’s success is one such achievement.
This is not the first time we have heard from DeepMind. Three years ago I wrote about AlphaGo, DeepMind’s game program that defeated world champion Lee Sedol in Go (https://www.angelesinvestments.com/insights/investment-insights/3rd-quarter-2017-ghost-moves). I noted that there are 10170 legal arrangements of the stones on a Go board, more than there are atoms in the universe. Like the placement of Go pieces, the number of protein shapes are too massive to crunch through every combination. And as with AlphaGo, AlphaFold found a shortcut. DeepMind took its champion gaming skills and applied them to unlocking the mysteries of biochemistry.
It’s not just a game; it is how our civilization advances.
Enjoy!
If you’d like to read more of Michael’s excellent blogs, on topics wide-ranging, please visit www.angelesinvestments.com
If you’d like to read more about the competition, bi-annual results, and the organization behind the project, please visit www.predictioncenter.org