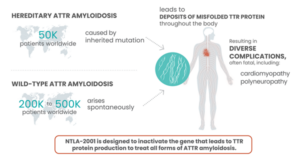
Amyloidosis is a group of diseases that have a common feature where proteins behave abnormally, with the breakdown products of these proteins folding upon themselves and depositing in various organs. Hereditary transthyretin amyloidosis is caused by a genetic mutation which causes misfolding of transthyretin (TTR) proteins (which originate from the liver). There are over 100 genetic variants of hereditary amyloidosis.
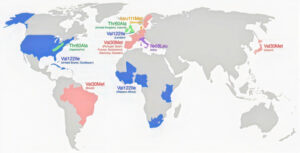
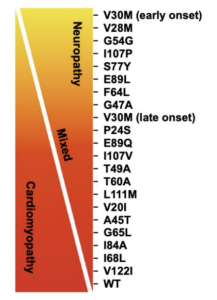
One such variant, called T60A, is the most common variant in Ireland (and the UK).
Symptoms of hereditary amyloidosis, specifically the T60A variant, include a variety of peripheral neuropathic, autonomic, and cardiac maladies, including:
- carpal tunnel syndrome
- numbness and tingling in hands, feet, arms, and legs
- muscular weakness
- excessive sweating
- dizziness/fainting (orthostatic hypotension)
- sexual disfunction
- unintentional weight loss
- indigestion
- acid reflux
- bouts of constipation and diarrhea
- fatigue
- shortness of breath
- leg swelling
- chest pain
One Patient’s Story
“Do you have an ancestor from Donegal?” is a question frequently asked by doctors who are investigating the possibility that a patient may have hereditary amyloidosis type T60A. With its origins in a short ribbon of coastline in North-West Donegal (Ireland), the condition wandered worldwide with Irish migration.”
(Callaghan, Donegal Amy, 2022.)
Donegal Ireland was one of the worst affected areas of Ireland’s “Great Hunger” of the mid to late 1800’s. 123,000 emigrants left the Donegal area between 1851-1900. A great many of them migrated to the United States, many to the Appalachian region of the country. The T60A variant, as it now appears in the United States, has been traced back to those settling in Appalachia. Sean Riley is a T60A amyloidosis patient who has ancestorial connections to Appalachia and the Donegal area.
Sean’s journey to diagnosis began in the fall of 2012 when he had bilateral carpal tunnel surgery. His job required quite a bit of typing and handwriting so he assumed that the condition was related to repetitive motion, which is a common cause of carpal tunnel syndrome. Little did he or the attending hand surgeon know that bilateral carpal tunnel syndrome may be an early neurological symptom of amyloidosis.
Concurrently, Sean started experiencing numbness in his left foot and lower left leg. He previously had vascular surgery on the left leg, and incorrectly assumed that the foot and leg numbness might be associated with nerve damage from the surgery. In actuality the numbness was due to the onset of peripheral neuropathy, yet another early symptom of the disease.
Between 2014 and 2017 he was taken to the hospital by ambulance on three separate occasions. In each instance he felt extreme dizziness and discomfort in his chest and assumed that the events were due to a cardiac issue, but no obvious signs of cardiac issues could be identified in any of the events. He now knows that what he was experiencing was orthostatic hypotension due to the onset of progressive autonomic neuropathy, another signature malady associated with the disease.
In the fall of 2017 Sean started being treated for severe acid reflux and gastrointestinal issues. Over time he had an endoscopy and colonoscopy performed, each which indicated normal results. These conditions likely indicated the onset of amyloidosis impact on nerves and tissue of the gastro-intestinal system.
Over the period of time from 2012 through 2017 Sean was seen by a hand surgeon, cardiology, oncology, endocrinology, neurology, and gastroenterology, along with his primary care physician. Nobody was able to connect the dots to amyloidosis, a product of the rarity of the disease and resulting lack of disease expertise by the general medical community.
In 2018 Sean moved overseas to Abu Dhabi to pursue a career opportunity. Shortly after arriving he started experiencing more frequent hypotensive episodes as well as progressive muscle wasting and weight loss. Fortunately for Sean the Cleveland Clinic has a hospital facility in Abu Dhabi. The attending cardiologist had a working knowledge of amyloidosis and ordered a series of tests, including an echocardiogram, a cardiac MRI, and a neuropathic evaluation, all of which concluded a preliminary positive diagnosis for the disease. As a result, the cardiologist recommended that Sean travel back to the United States and be seen at the amyloidosis center at Brigham and Women’s hospital in Boston. In February of 2019 he received a definitive diagnosis of hereditary transthyretin amyloidosis, specifically the T60A mutation. Excerpts of the confirming echocardiogram, cardiac MRI, and genetic testing results are shown below.
Echocardiogram Summary Notes

Associated Cardiac MRI Interpretation

DNA Sequencing Result

Shortly after diagnosis, Sean started treatment with a state-of-the-art FDA-approved amyloidosis drug. The treatment is administered every three weeks and is designed to slow or stop disease progression. The drug is an RNA signal blocker which stops the transthyretin proteins from misfolding and creating amyloid fibrils.
Sean continues this therapy to this day, and all indications show that disease progression has stopped. There is no cure for the disease, so he must contend with and manage the damage that has been done; however, he is thrilled that the disease progression is being kept in check.
For more information on hereditary amyloidosis worldwide, visit our blog — Click Here
Bibliography
“Donegal Amy-A Rare Inherited Disease from Ireland”, Rosaline Callaghan, Roscara Books, 2022.
“Unraveling the Lineage: The Genetic Basis of Familial ATTR Cardiomyopathy Ronald Witteles”, MD Professor of Medicine (Cardiovascular Medicine).
“Cardiac Amyloidosis Part 1: Understanding Types and Risks”, Dr. Rodney Falk, Brigham and Women’s Hospital, YouTube, July 2018.