Cardiomyopathy is a broad term that is used to describe disease of the heart muscle, making it difficult for the heart to provide the body with an adequate blood supply. It can lead to heart failure and even death. In this article, we’ll discuss the types of cardiomyopathy and its connection to amyloidosis.
Risk Factors
It has no ideal target, as it can affect a person of any age, race, or gender. However, there are a number of risk factors that can put one at an increased chance of developing cardiomyopathy.
- Genetic History → Family history of cardiomyopathy, heart failure, or sudden cardiac arrest
- High Blood Pressure → Over a long period of time
- Heart Conditions → Past history of heart attack, coronary artery disease, or infection of the heart
- Obesity → Tends to make the heart work harder to perform its normal function
- Alcohol Use → Long period of alcohol use
- Drug Use → Use of illicit drugs, such as cocaine, amphetamines, and anabolic steroids
- Medications → Drugs used in the treatment of cancer, such as chemotherapy and radiation
Additionally, there are a number of diseases that increase the risk of developing cardiomyopathy, including:
- Amyloidosis
- Connective Tissue Disorders
- Diabetes
- Hemochromatosis (excess iron storage)
- Sarcoidosis
- Thyroid Disease
Types of Cardiomyopathy
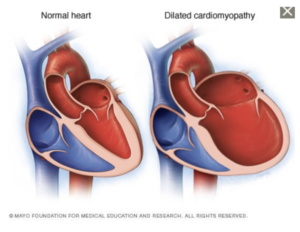
- Dilated Cardiomyopathy → Dilation of the left ventricle prevents the heart from pumping effectively. It most commonly occurs in middle-aged men and is typically the result of coronary artery disease, heart attack, or genetic defects.
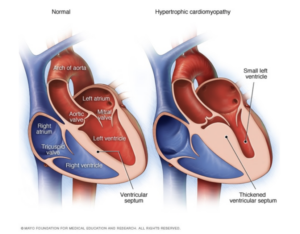
- Hypertrophic Cardiomyopathy → Abnormal thickening of heart muscle, most commonly affecting the muscles surrounding the left ventricle. This type of cardiomyopathy is strongly associated with a family history of the disease. There have been genetic mutations linked specifically with this type of cardiomyopathy.
- Restrictive Cardiomyopathy → Stiffening of the heart muscle results in an inelasticity, making it difficult for the heart to expand and fill. It is most commonly seen in the elder population. The disease can be of idiopathic origin or of disease such as amyloidosis. This is the least common type of cardiomyopathy.
- Arrhythmogenic Right Ventricular Dysplasia → Scar tissue replaces healthy tissue of the right ventricle. This form of cardiomyopathy is rare and often the result of genetic mutations.
- Unclassified Cardiomyopathy → All other forms of cardiomyopathy fall within this category.
Amyloidosis
Cardiomyopathy is one of the hallmarks of amyloidosis, often seen in the transthyretin form of amyloidosis (ATTR). ATTR-CM, or transthyretin amyloid cardiomyopathy, is a disease where the transthyretin protein becomes unstable and misfolds. This unstable protein (“amyloid”) then deposits in the heart muscle, resulting in thickening and stiffening of the heart.
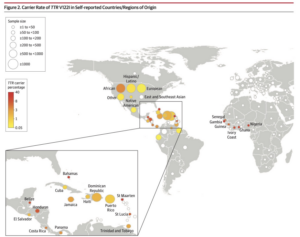
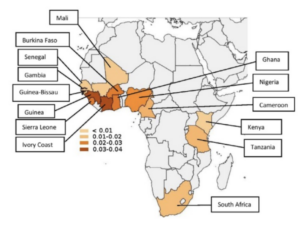
The two types of ATTR-CM are wild-type ATTR-CM (wtATTR) or hereditary ATTR-CM (hATTR). wtATTR-CM is the most common form of ATTR-CM, affecting predominantly white males 60+ years old. hATTR-CM is genetic affecting both men and women, and presents as early as 50+ years old. Interestingly, one of the mutations causing hATTR, V122I, is seen almost exclusively in individuals of African ancestry. It is believed that approximately 3-4% of African Americans carry this mutation, regardless of whether or not they develop symptoms.
Most importantly, these are the most common and important signs and symptoms to be aware of, in order to diagnose ATTR amyloidosis.
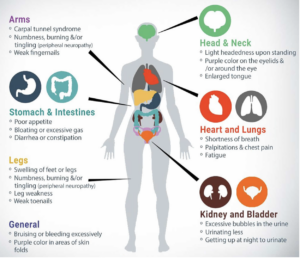
Expert Insights – Cardiac Clues and Clinical Signs
In part 1 of a 2-part series, Dr. Keyur Shah, cardiologist from VCU Health’s cardiac amyloidosis care team, discusses the two most common types of transthyretin (TTR) amyloidosis: hereditary and wild-type. He details how ATTR cardiomyopathy amyloidosis presents and manifests itself to impair the heart. Dr. Shah lists clinical clues, “red flags,” and biomarkers which can raise suspicion of the presence of amyloidosis. Next he discusses insights that can be gained from echocardiograms, electrocardiograms, and cardiac MRIs and how they offer possible indicators of the disease presence. Once amyloidosis is suspected, definitive diagnosis testing is next.
In part 2 of a 2-part series, Sarah Paciulli, Heart Failure Nurse Practitioner, from VCU Health’s cardiac amyloidosis care team, continues from where Dr. Keyur Shah ended in Part I and discusses here in Part II the non-cardiac clues of transthyretin (TTR) amyloidosis. She expands the list of clinical clues and “red flags” that clinicians should be alert to, including orthopedic manifestations, erectile dysfunction, and polyneuropathy.
===========================================================
References:
https://www.mayoclinic.org/diseases-conditions/cardiomyopathy/symptoms-causes/syc-20370709
https://www.yourheartsmessage.com
https://healthjade.net/familial-amyloidosis/