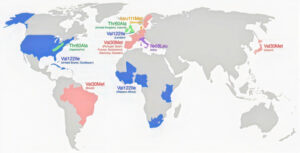
Transthyretin Amyloidosis, or ATTR, is considered a single disease, however the diversity in its clinical presentation is staggering. In this blog, we’ll discuss some of the most common hereditary variants and how the disease manifestation differs around the world in documented hotspots.
Source: Epidemiology, Genetics, and Prognostic Factors (1)
There are two distinct forms of Transthyretin Amyloidosis (ATTR), the hereditary form (ATTRv), and the non-hereditary form (ATTR-wt) commonly referred to as wild-type amyloidosis. Disease manifestation is considered a spectrum involving aspects of cardiomyopathy, neuropathy, or more frequently a mixture of both.
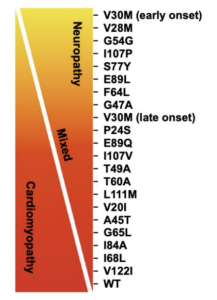
Below we’ll discuss the hereditary form and the various genetic variants and how they differ based on geographical location.
WHAT IS TRANSTHYRETIN (TTR)
Transthyretin, also known as prealbumin, is a protein produced primarily in the liver that is responsible for the transport of thyroxine and retinol. Interesting enough, this is how it got its name.
In steady state, the protein circulates primarily as a tetramer (i.e., monomeric form), but unfortunately, its monomeric form is inherently amyloidogenic (prone to breakdown and formation of amyloid aggregates). Couple that with mutations that increase the amyloidogenicity of the protein, these tetramers dissociate into monomers that will misfold, aggregate, and form the insoluble fibrils (“amyloid”) that are resistant to the body’s inherent protective mechanisms like proteolysis.
SPECIFIC TTR PATHOGENIC VARIANTS
As of today, there have been over 145 reported variants related to hereditary transthyretin amyloidosis. Interestingly, these genetic variants have a tendency to cluster in both geographic and ethnic groups around the world. We’ll discuss some of the most prevalent mutations below.
Val122Ile
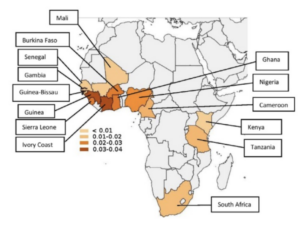
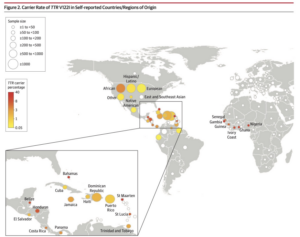
This is the most common TTR mutation in the United States, with a prevalence of roughly 3.4% in the African American community. The disease is primarily cardiac in nature, typically present when patients are in their 60s. It is thought that this mutation arose from the region of West Africa and has worked its way to the United States over time, where it has become the predominant form.
Val30Met
This is the most commonly recognized TTR mutation worldwide and the first TTR variant discovered. It is most commonly found in the regions of Portugal, Spain, France, Japan, Sweden, and Brazil. Interestingly, between these regions where this mutation dominates, there is variability in age of onset and parent-of-origin. For example, age of onset was found to be earlier in the Swedish population in comparison to Portugal and Japan. As for the parent-of-origin, it was found that the mother was more likely than the father to pass along the mutation (153 vs. 138), whereas in the French population the father was more likely to pass on the mutation (219 vs. 216), although not by much. The one thing these populations do have in common is this form of the disease is almost exclusively neurologic in nature.
Thr60Ala
This variant is most commonly found in the UK and Irish populations, and is also seen in the Appalachian region of the United States. This variant presents as a mixture of both cardiomyopathy and neuropathy symptoms. It seems to be that in early stages of the disease the neurologic symptoms are most prevalent, but cardiac symptoms present at diagnosis seem to indicate poorer patient outcomes.
Thr119Met
This is arguably the most interesting variant that was investigated in a large study of the Danish population. The presence of this mutation actually confers a protective benefit. When this mutation occurs along with the Val30Met mutation, it has the effect of stabilizing and delaying, even preventing transthyretin amyloidosis.
PROGNOSIS
While the prognosis is by no means near perfect, it is improving. There is continued advancement in the field of transthyretin amyloidosis, whether it be improving diagnostic methods, drug development, or a potential cure on the horizon with CRISPR gene-editing technology. Having said that, there continue to be significant barriers to diagnosis. The importance of being an astute clinician to suspect and work up for amyloidosis remains at the forefront of the challenge.
CONCLUSION
The geographic nature of this disease plays an important role in identifying and diagnosing amyloidosis. Having an understanding of how the presentation of the disease is heavily related to the patient’s ancestry and location around the world. The hardest part is suspecting amyloidosis, from there don’t forget the value of the diagnostic tools at your disposal, including genetic testing. Use this knowledge to strengthen and guide your suspicions of amyloidosis!
Over the upcoming months we’ll post blogs delving deeper into some of these variants, so stay tuned.
Thanks for reading,
Mackenzie
————————————————–
SOURCES